
Síndrome de Prader-Willi: Una enfermedad genética extraña
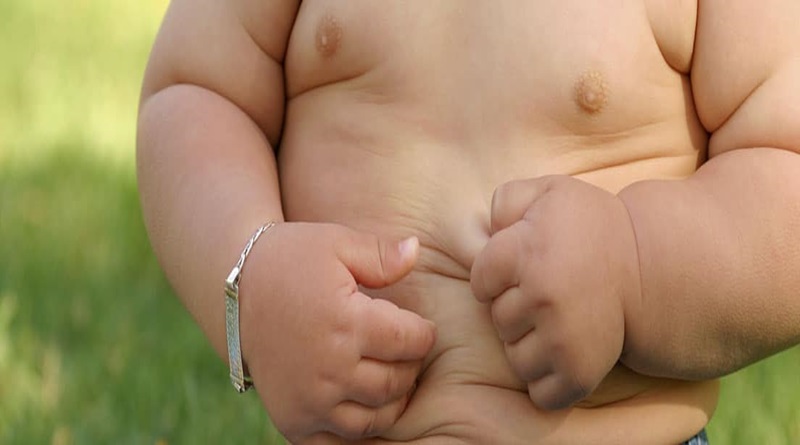
Cada 30 de mayo, se conmemora el síndrome de Prader-Willi (SPW), una enfermedad genética extraña que afecta aproximadamente a 1 de cada 15.000 personas. Se caracteriza por una variedad de síntomas, incluyendo baja tonicidad muscular, retraso en el desarrollo, problemas de comportamiento y constantes sensaciones de hambre que pueden llevar a la obesidad si no se controla adecuadamente.
A pesar de que el origen del síndrome de Prader-Willi es genético y no puede prevenirse, existen medidas que pueden tomarse para controlar los síntomas y mejorar la calidad de vida de las personas afectadas. El tratamiento suele incluir terapias físicas y ocupacionales, así como una dieta controlada y supervisada por profesionales de la salud.
El síndrome de Prader-Willi fue descrito por los Dres. A. Prader, H. Willi y A. Labhart en el año 1956, aunque hay noticias del síndrome en toda la historia (existe un cuadro de 1680 en el Museo del Prado de Madrid, que retrata una joven PW).
No hay razones conocidas para este accidente genético complejo que afecta el apetito, crecimiento, metabolismo y las funciones cognoscitivas. Tienen bajo tono muscular y presentan problemas de salud y de comportamiento característicos. La característica principal es una necesidad involuntaria de comer constantemente, la cual, unida a una necesidad de calorías reducidas, lleva a la obesidad.
Aunque el SPW se asocia a una anomalía en el cromosoma 15, no está considerada como una condición hereditaria, sino un defecto genético espontáneo que se da durante o en un momento cercano a la concepción. El SPW se encuentra en personas de ambos sexos y en todas las razas.
Es una anormalidad (deleción intersticial del 15q.) congénita, detectable (50%) en el par masculino del cromosoma número 15, no hereditaria (recurrencia >0.1%), resulta condición de por vida, peligrosa para la supervivencia del afectado, presente en cualquier raza sin distinción de género sexual, con una frecuencia estimada de 1/10.000 nacimientos.
De difícil diagnóstico neonatal (salvo análisis prometafase de alta resolución positivo), el afectado se presenta tras escaso movimiento intrauterino y parto normal, con bajo peso, hipotonía, incapacidad para mantener la postura en cabeza y extremidades, llanto escaso y débil, pobres reflejos al succionar, fatiga, dificultad en secreciones, letargia, síndrome de ventilación reducida, cambios poco pronunciados en la apariencia física, hipogenitalismo y apetito escaso, siendo necesarios cuidados intensivos (90%) y alimentación asistida.
De adulto, con características clásicas (89%): adiposis extrema (95%) en tronco inferior y muslos por apetito insaciable, comer incontrolado, estatura corta (>152cm.) por deficiencia de la hormona de crecimiento, medida pericraneal, manos y pies pequeños, disfunciones del sistema nervioso, no desarrollo de las características sexuales (criptorquidia y/o hernias inguinales, hipogonadismo e hipogenitalismo, amenorrea, infertilidad), poca sensibilidad al dolor, variaciones en la temperatura corporal, inhabilidad para el vómito (66%), debilidad de la musculatura principal, débil coordinación, falta de equilibrio, cansancio, excesivo deseo de dormir; motricidad general, desarrollo motriz, funciones motoras, lenguaje, articulación verbal y etapas de desarrollo lentos (típicamente entre 1-5 años de edad), saliva pegajosa, debilidad de la superficie dental, osteoporosis y envejecimiento prematuro.
Con coeficiente intelectual (CI) >70 puntos (variación 40-105%), retraso mental (90%) (oligofrenia, amencia), pensamiento abstracto y comprensión de conceptos débil, problemas de conducta (obstinación violenta), tensión familiar, obsesión con las heridas en la piel, depresión y episodios psicopáticos en la edad preescolar, agravados con la pubertad e insaciable deseo de comer. Frecuentemente, estrabismo, miopía, escoliosis temprana y diabetes II con resultado fatal.
Sin medicación específica conocida y difícil de adaptarse a la sintomatológica, los efectos del síndrome pueden reducirse, dependiendo de los medios disponibles, en un ambiente familiar controlado, con atención temprana, enseñanza especial, dirección psicológica, sociológica, fisioterapéutica, logopédica y endocrinológica adecuadas y, sobre todo, mediante un estricto control del peso. Aun así, los problemas persisten durante toda la vida y es muy raro que los individuos afectados logren una vida independiente con éxito.
En el Día Internacional del Síndrome de Prader-Willi, es importante concienciar sobre la importancia de la prevención y el tratamiento adecuado de esta enfermedad. Se debe promover la investigación y el apoyo a las personas afectadas, así como educar a la sociedad sobre la importancia de la inclusión y la empatía hacia quienes viven con esta condición.
Fuente: Medios Digitales
VTV/DR/CP


